2025年9月9日,我校特聘教授陈实富博士,在iMeta(中科院1区,IF=33.2),以独立作者发表高性能生物信息学软件fastp1.0(我校为第一单位)。该生物信息学软件因其强大的功能和极致的性能而被公认为最受欢迎的 FASTQ 文件预处理工具之一,已经被引用了2万多次。作为其首次重大更新,本文正式介绍 fastp 1.0 的新功能及其背后的实现原理。为了展示fastp在简洁性、高效性和多功能性方面的巨大优势。
测序技术是探索生命科学和进行分子诊断的核心技术。由于测序数据复杂且易出错,通常需经过质量控制和清洗,才能进入下游分析。预处理步骤通常包括对原始测序数据进行质量统计、去除接头、过滤低质量序列或碱基,并修正部分测序错误。研究表明,数据预处理对于获得可靠的分析结果至关重要。
鉴于数据质量控制和预处理的重要性,研究人员已开发出多种相关工具。其中,Trimmomatic、Cutadapt 和 fastp 是使用最广泛的三种。Cutadapt 于 2011 年首次发布,正值高通量测序技术迅猛发展之际,之后持续更新,广泛用于接头和引物序列的去除。然而,Cutadapt 在质量控制和数据过滤方面相对薄弱,且缺乏一些现代功能,如接头自动检测、数据拆分与合并、UMI(唯一分子标识符)处理等。Trimmomatic 于 2014 年发布,最初主要用于去除 Illumina 接头序列,后续逐渐增加了修剪和质量过滤功能。然而,Trimmomatic 在过去五年中未再更新,缺少处理 UMI 或执行去重等关键功能。此外,Trimmomatic 和 Cutadapt 无法在去除接头和过滤数据的同时,完成全面的数据质量统计分析。这通常要求用户在使用这些工具前后,分别运行 FastQC 或 MultiQC 来评估数据质量的改善,导致计算资源的显著浪费和数据分析成本的增加。
作为后起之秀,fastp 于 2018 年首次发布,并持续开发和迭代。在 fastp 的设计初期,就充分考虑了如何最大限度地降低成本,既包括学习成本,也包括计算成本。因此,fastp 能够在完成数据过滤和修剪的同时,对预处理前后的数据进行全面质量分析。此外,fastp 的算法经过高度优化,即使功能更丰富,其运行速度仍远超 Trimmomatic 和 Cutadapt。同时,fastp 专为云端使用场景设计,内存占用极低,有效降低云服务器成本,并提供丰富的 HTML 报告,便于通过网页端查看。
fastp 是一个多线程、多功能的 FASTQ 数据预处理工具,遵循简洁、高效、多功能和可重现四大设计原则。无论输入是单端还是双端数据,使用何种参数,其多线程队列机制都能确保结果稳定且可重现。
简洁并不意味着功能减少或能力变弱,而是指在隐藏实现细节的同时,提供一个易于理解的界面。所谓简洁,就是预判绝大多数用户的需求,并为他们默认做出最优选择。fastp 已经历了近 50 次小版本迭代,但始终恪守简洁原则。fastp 的简洁体现在多个方面:例如,它的默认参数极其简单,只需指定输入和输出文件即可,且默认模式已足够强大,能满足绝大多数常见场景;再如,大多数情况下用户根本无需手动输入接头序列,因为 fastp 会自动检测——尽管背后的算法可能非常复杂,但用户无需关心这些细节。
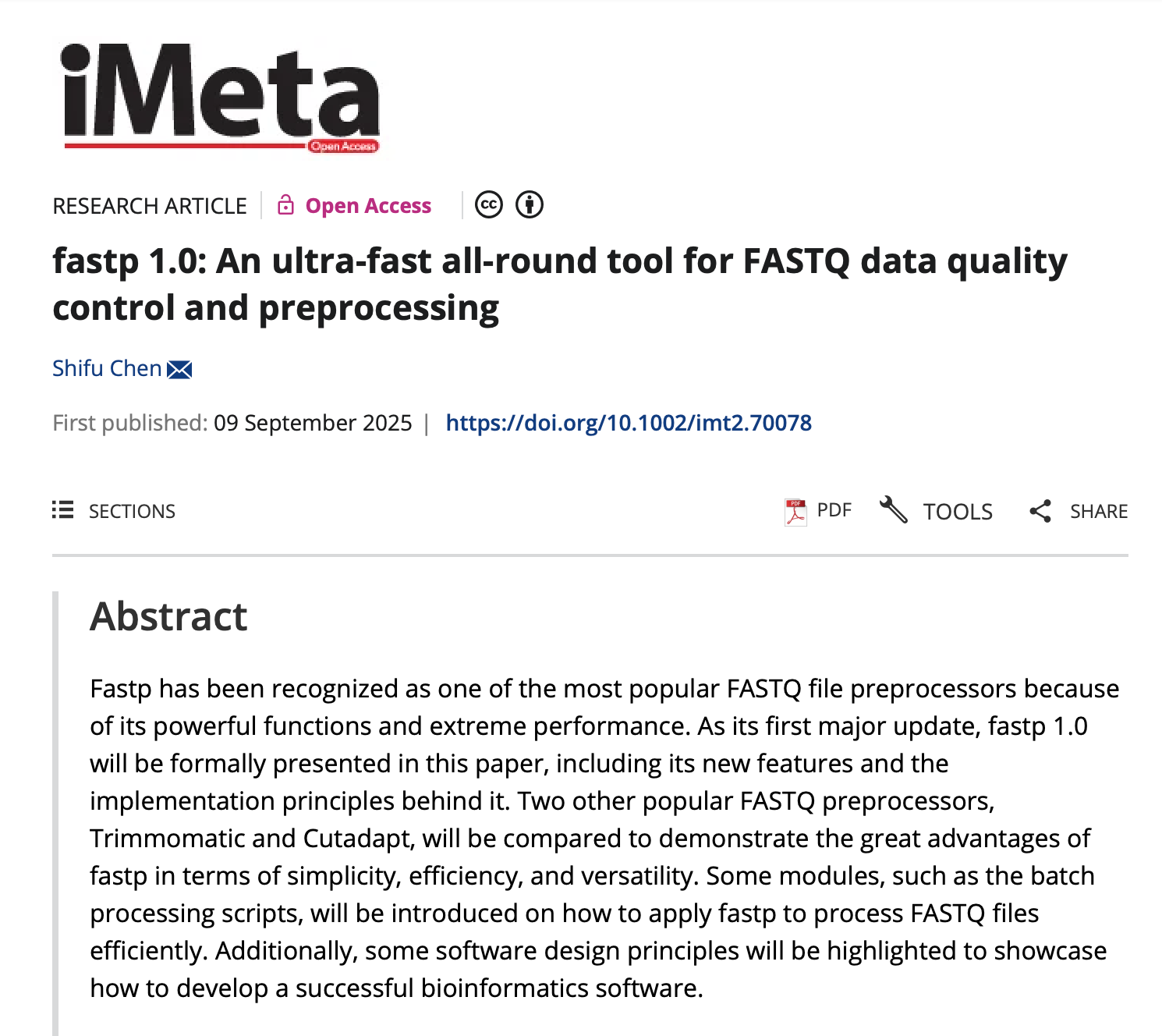
fastp 简洁性的另一个体现是其 HTML 报告。与纯文本或 CSV 报告相比,HTML 显然更加用户友好,尤其适合云端环境。在 fastp 1.0 中,HTML 报告的最大改进是提供了预处理前后数据的并排对比。
预处理前后数据质量的并排对比
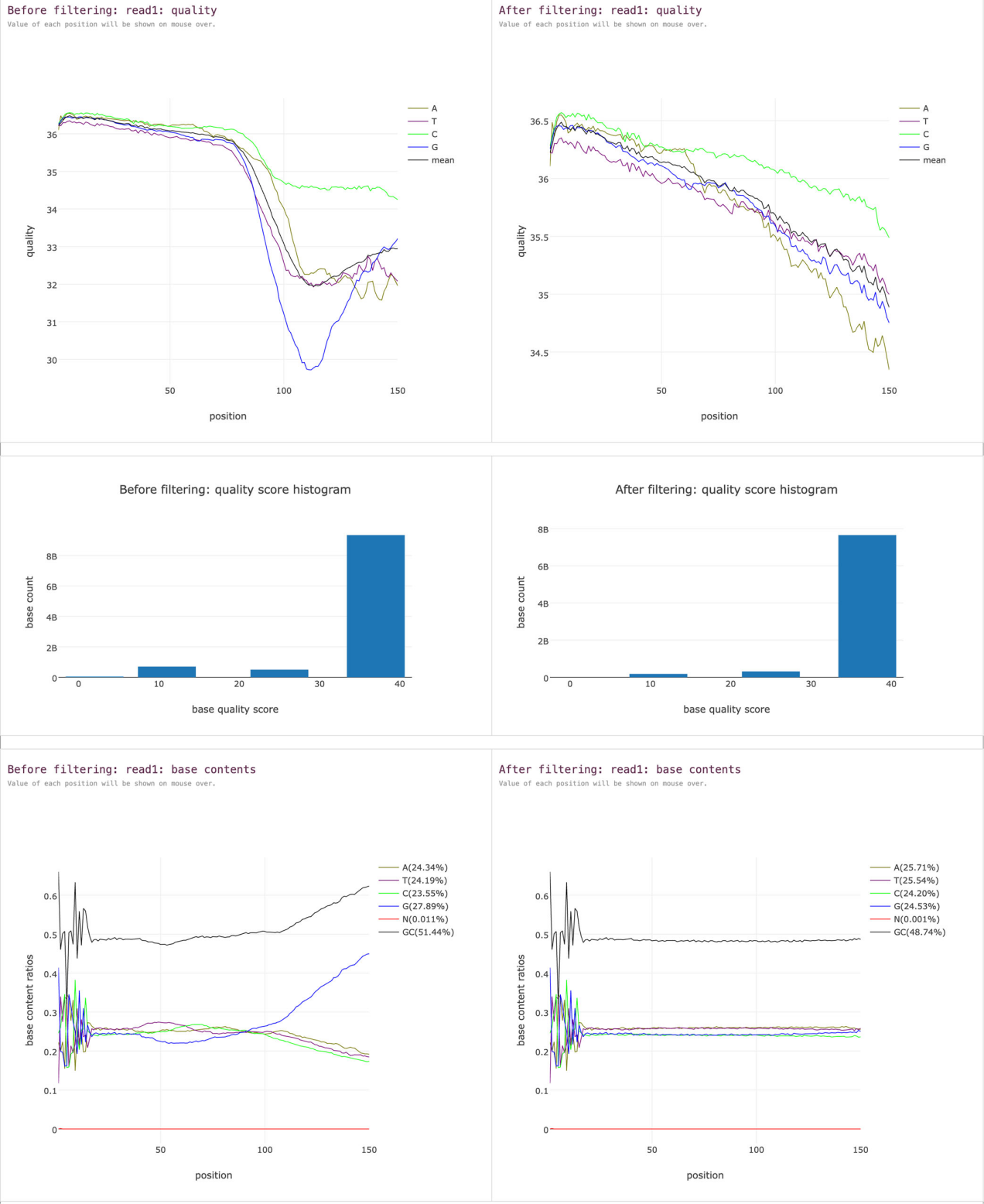
fastp 1.0 的简洁性进一步提升:提供了高效脚本以并行批处理大量 FASTQ 文件。该脚本会自动对文件夹内的所有 FASTQ 文件进行预处理,并智能配对双端数据,最终生成一份汇总 HTML 报告,展示所有样本的总体统计信息。下图展示了 10 个 FASTQ 数据经并行处理后生成的汇总报告示例。
由 fastp 并行批处理脚本生成的 10 个单端 FASTQ 文件汇总报告。所有样本的数据质量均获得大幅提升。
高效通常包含两层含义:高速度与低计算成本,这两点对于生物信息学分析都至关重要。高通量测序文件通常体积极大,处理耗时长,因此计算速度尤为关键;同时,如今绝大多数生信流程都在云端运行,软件必须尽量降低 CPU 和内存需求,以节省开销。
为了提升 fastp 的计算速度并减少资源消耗,作者做了大量优化。例如,fastp 采用了“边过滤边质控”的并发模式:只需一次读取即可完成数据修剪、过滤,并获得预处理前后的全部质量统计结果。
在检测接头或引物序列时,经常需要允许序列比对时存在空位(gap-tolerant)。对于短序列,为保证精度通常仅允许 1 个 gap,此时若采用传统序列比对算法或编辑距离计算,速度会非常慢,带来巨大开销。fastp 1.0 为此设计了一种“单 gap 匹配算法”,显著加速该过程。该算法简洁而高效,将计算复杂度从 O(n²) 降至 O(n)。
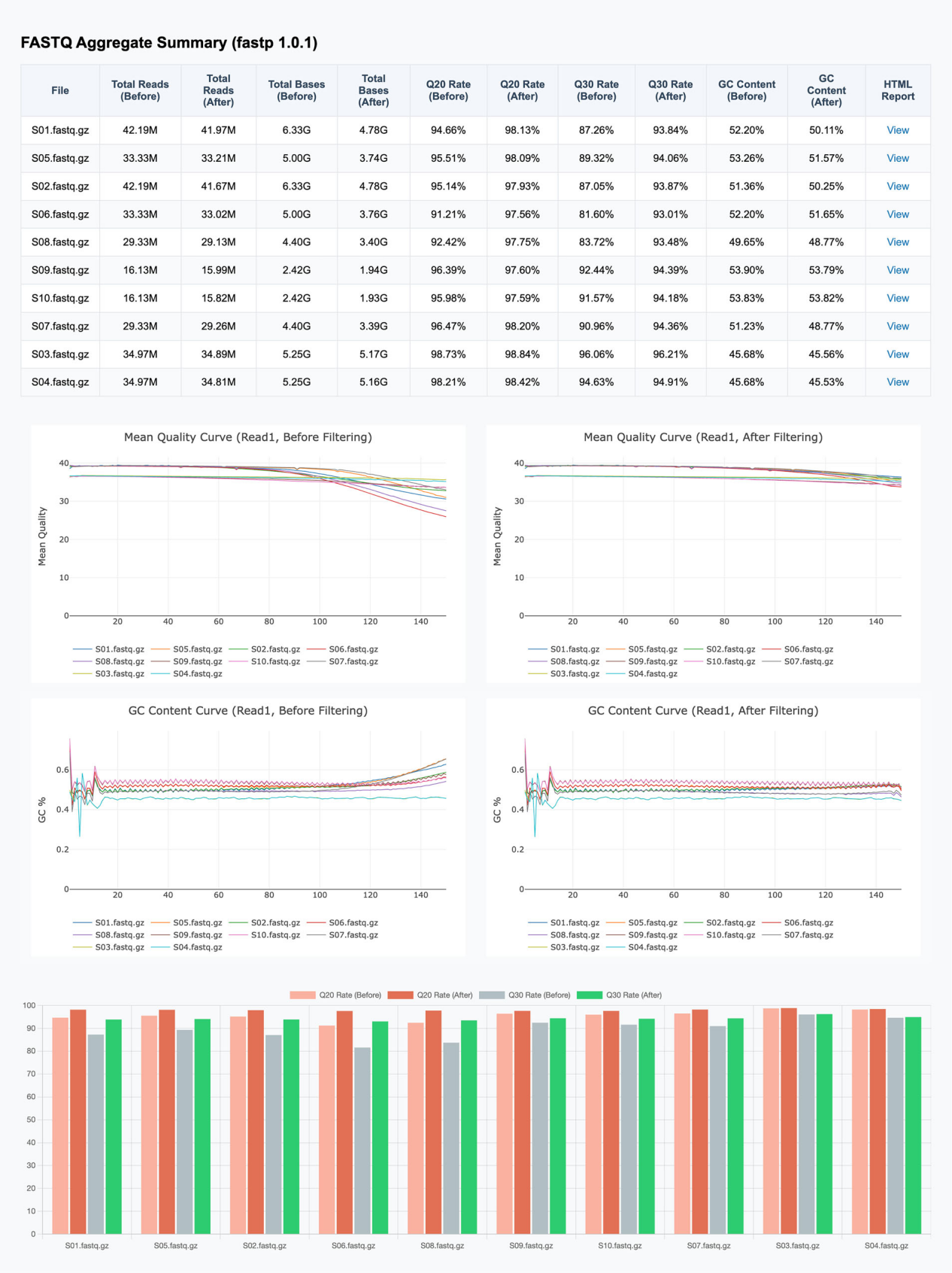
由于测序技术的广泛应用,数据预处理的需求也极为多样,这就要求预处理工具具备丰富的功能。表 1 列出了 Trimmomatic、Cutadapt 与 fastp 的功能对比,所有信息均来自各软件的官方文档。可以看出,fastp 已涵盖了绝大多数必需的预处理功能(见表 1)。当然,Trimmomatic 和 Cutadapt 也都是优秀的工具,在某些场景下它们甚至可以超越 fastp。例如,Cutadapt 提供了 demultiplexing(样本拆分)功能,而 fastp 并未实现——因为作者认为这应由专门的 demultiplexer 完成。为了保持简洁,fastp 刻意省略了一些关联度较低的功能。
表1. fastp、Trimmomatic 与 Cutadapt 功能对比

可重现性是生物信息学软件开发中常被忽视却至关重要的一项原则。由于生物信息学软件本身复杂且多样,大多数用户难以深入理解其内部细节,因此只能依赖软件输出的稳定性来获得可重复的结果。Fastp 1.0 在任何输入输出、启用的模块或所用参数下,均能保证结果的可重现性。
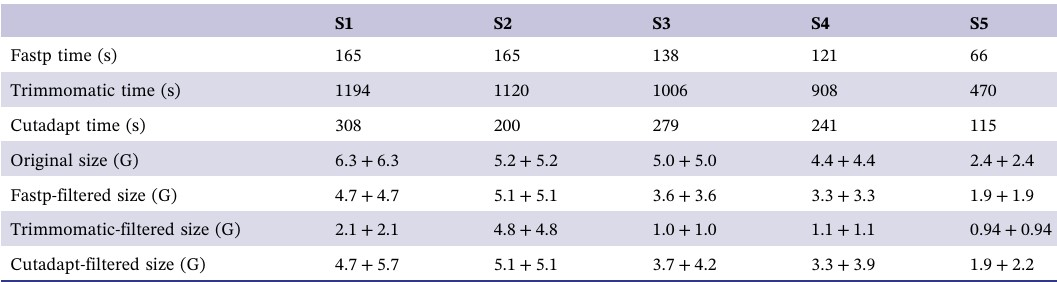
Fastp 的超高效率是其在生物信息学界被广泛应用的主要原因之一。为验证这一点,作者开展了一项性能评估实验,比较了 Trimmomatic、Cutadapt 和 fastp 处理同一批数据所耗时间(见表 2)。
表2. fastp、Trimmomatic 和 Cutadapt 的速度评估
文献来源
Chen, Shifu. 2025. “fastp1.0: An Ultra‐Fast All‐Round Tool for FASTQ Data Quality Control and Preprocessing.” iMeta e70078.https://doi.org/10.1002/imt2.70078
作者简介

陈实富,博士,赣南医科大学特聘教授,海普洛斯联合创始人。连续多年入选全球前2%顶尖科学家榜单(肿瘤学和生物信息学方向)和中国高被引作者(计算机科学方向)。发起开源项目组OpenGene,开发了超过10款热门生物信息学软件,在STTT, iMeta, Nat Comm, Can Res, BIB, Bioinformatics等著名期刊发表论文60余篇,最高单篇引用2万多次。在生物信息学和肿瘤标志物领域拥有数十项发明专利和软件著作权。任高水平国际期刊《iMeta》(IF=33.2) 执行副主编,中国抗癌协会基因诊断专委会委员,中国工业与应用数学学会“数学与产业专业委员会”委员。


